|
|
|
| Click here to visit our web site |
|
INTRODUCTION Pathologic increases in osteoclast differentiation and activation occur in experimental models of osteolytic metastases(4), primary bone tumors(5), humoral hypercalcemia of malignancy(6,7), osteoporosis(8), rheumatoid arthritis(9), and wear-induced periprosthetic osteolysis(10-13). In this review we will discuss the recent developments in the application of the novel therapies Osteoprotegerin and RANK.Fc to these osteolytic disease processes.
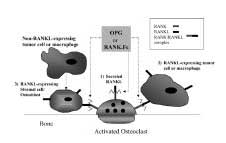
RANK and RANKL RANK is a 616-amino acid, type I membrane protein in the tumor necrosis factor receptor (TNFR) family. It contains four extracellular cysteine-rich pseudorepeats(2). It is expressed in osteoclasts, pre-osteoclasts(3), and several malignant cell types(16). RANKL (RANK Ligand), also known as OPGL (osteoprotegerin ligand)(17), ODF (osteoclast differentiation fac-tor)(3) and TRANCE(14), is the obligate ligand for RANK. (2) It exists naturally in membrane-bound2 and secreted forms(18-20), and is expressed in stromal fibroblasts, osteoblasts(3), T cells(2), and malignant cells(5, 20, 21). Osteoblast expression of RANKL is strongly stimulated by several osteolysis-inducing agents including (1, 25) (OH) (2) vitamin D(3), PTH (and PTHrp), IL-6 and IL-11(3). Interaction between RANKL-expressing cells and RANK-expressing pre-osteoclasts triggers an intracellular signaling cascade, which via TNFR associated factors (TRAF) 1-6, effects the NF-kB, serine/threonine kinase Akt/PKB, and protein kinase c-Jun N terminal kinase (JNK) pathways(22-27). How these pathways regulate osteoclast activity has yet to be clearly defined. OSTEOPROTEGERIN (OPG) RANK.Fc PRE-CLINICAL STUDIES OF OSTEOPROTEGERIN AND
RANK.Fc Although the majority of prostatic carcinoma
metastases in bone are clinically osteosclerotic (osteoblastic) rather
than overtly osteolytic, OPG inhibits
prostate carcinoma growth in bone. Cultured
prostatic carcinoma cells produce a soluble form of RANKL, and directly
stimulate osteoclastogenesis in the absence of underlying stroma. In mice,
OPG treatment completely inhibits osteoclastogenesis and the formation
of mixed osteoblastic/osteolytic skeletal tumors(20).
However, serum OPG levels(31)
and OPG/RANKL mRNA-isolate ratios are elevated in patients with advanced,
metastatic prostate cancer. This seeming contradiction may be explained
by the observation that prostate metastases often begin with cycles of
osteolysis, which progress to an osteoblastic reaction in later stages.
OPG may inhibit the early osteolytic component of prostatic metastasis,
thus preventing the destruction of bone requisite for the establishment
and neovascularization of metastatic foci. This emphasizes the significance
of OPG/RANKL concentration ratios in the development of lytic versus blastic
type lesions. Local OPG/RANKL ratios may initially be low, but increase
once metastases have been established. Giant cell tumor (GCT) stromal cells contain high levels of RANKL mRNA, while giant cells themselves exclusively express RANK mRNA. GCT tissues express relatively higher levels of RANKL mRNA in comparison to OPG mRNA in vitro. OPG dose-dependently inhibits osteoclast formation and bone resorbing activity in GCT of bone in vitro. This process is reversed by treatment with recombinant RANKL. In GCT, osteoclast differentiation can be stimulated directly by RANKL expressing tumor stromal cells, as is seen in prostate cancer(5). This contrasts the proposed indirect induction of osteoclastogenesis seen in breast cancer metastases and multiple myeloma. Serum OPG levels are lower in patients with multiple myeloma (MM), and correlate inversely with the severity of osteolytic disease(32). RANKL and OPG mRNA production are up- and down-regulated, respectively, in cultured bone marrow explants of patients with MM in comparison with healthy controls(33). Myeloma cells stimulate and inhibit RANKL and OPG production, respectively, in co-culture studies(34). OPG administration in a mouse model was effective in preventing the formation of osteolytic lesions, and was associated with decreased osteoclast formation and increased bone density(35). Osteoclastogenesis was inhibited by the addition of RANK.Fc to co-cultures, and RANK.Fc prevented myeloma-induced bone destruction and slowed disease progression in a SCID-hu murine model of human myeloma(34). OPG blocks, and actually reverses bone cancer pain-related neurochemical reorganization in the spinal cord of mice(36, 37). These studies strongly suggest the potential use of OPG in the treatment of bone cancer- related pain. OPG and RANK.Fc have also been evaluated in humoral hypercalcemia of malignancy. Mice with induced colon-26 cancer have increased PTHrp (parathyroid hormone relatedpeptide) expression, elevated serum PTHrp, increased bone resorption, and marked hypercalcemia. OPG treatment initiated at the onset of hypercalcemia, or after it occurs, results in inhibition of tumor-induced bone resorption and hypercalcemia, and subsequent normalization of serum ionized-calcium levels(6). RANK.Fc effectively inhibits PTHrp-induced resorption in bone cultures in vitro. Administration of murine RANK-human Fc fusion protein to normal mice resulted in the disappearance of osteoclasts from the metaphyses of long bones and increased calcification of bony trabeculae as seen radiographically(7). In nude mice with subcutaneously implanted human lung cancer tissue, RANK.Fc also inhibits osteoclastic bone resorption and hypercalcemia, without affecting circulating levels of PTHrp. The potential therapeutic value of RANK.Fc is strengthened by its ability to reverse the increase in blood ionized-calcium level even after hypercalcemia had been established in this model7. ARTERIOSCLEROSIS RHEUMATOID ARTHRITIS PERIPROSTHETIC OSTEOLYSIS An in vitro mouse study suggests that OPG might be effective in treating wear-induced periprosthetic loosening. Kim et al. were able to demonstrate increased osteoclast formation in a co-culture system in the presence of failedarthroplasty joint fluid compared with OA joint fluid. They also demonstrated lower OPG levels in the failed-arthroplasty fluid. The increased osteoclastogenesis was blocked by administration of exogenous OPG39. Childs et al. analyzed effects of RANK.Fc on titanium –induced osteolysis in a mouse calvaria model, making several provocative observations. First, titanium–induced osteoclastogenesis and bone resorption was blocked by intraperitoneal doses of RANK.Fc greater than 1mg/kg, given every 48 hours. A 10mg/kg dose completely inhibited osteolysis. Interestingly, these data were statistically equivalent to titanium implants in RANK-knockout (RANK -/-) mice. Second, mice treated with a single dose 5 days following implantation were depleted of TRAP+ (tartrate-resistant acid phosphatasepositive) cells 16 days later. Third, the significant bone loss during the first 5 days was restored by day 21. These studies suggest that inhibition of RANK prevents and reverses wear debris-induced osteolysis without affecting osteogenesis. RANK.Fc has not been tested in humans to date(40). OSTEOPROTEGERIN IN PHASE II CLINICAL TRIAL
DISCUSSION Future directions for this research should include inquiries into endogenous OPG, RANK, and RANKL: cell sources and gene regulation, modes of sequestration, inhibition, specificity, activation, proteolytic cleavage, and clearance. For example, serum OPG levels may eventually be correlated with disease prevalence, risk of skeletal metastasis or implant failure. Elucidation of the human genetics of the RANKL/RANK/OPG system, including the specific downstream signaling molecules in different cell types, will be essential for understanding and treating a broad range of inherited and idiopathic skeletal diseases. Familial Expansile Osteolysis is now known to be an autosomal dominant disease caused by a mutation in the gene for RANK(42, 43). Perhaps mutations in the genes encoding RANKL or OPG are the cause of other yet-unexplained diseases. Finally, the RANKL/RANK/OPG system is an attractive target for small molecule drug design which may generate additional therapeutic options. Martin M. Dolan, MD is a Resident in the Department of Orthopaedic Surgery at the Hospital for Special Surgery and a Research Fellow in the Department of Orthopedic Surgery, Children’s Hospital. Peter V. Hauschka, PhD is Associate Professor of Orthopaedic Surgery at Harvard Medical School, Associate Professor of Oral Biology at Harvard School of Dental Medicine, and Senior Research Associate in Orthopaedic Surgery at Children’s Hospital. Address correspondence to: |
|
Print Manuscript • View References • Download PDF version • Close window |