|
|
|
| Click here to visit our web site |
|
INTRODUCTION The discovery in our laboratory that chondrocytes express IL-1 genes raised the possibility that IL-1s produced by chondrocytes, rather than by synovial cells, play a role in OA pathogenesis. Subsequent studies from this laboratory have demonstrated that IL-1 is expressed in OA cartilage and that IL-1 proteins accumulate within chondrocytes early in the development of OA.(2-5) The concept that IL-1s are important in OA pathogenesis is further supported by work showing that the natural IL-1 inhibitor IL-1receptor antagonist (IL-1ra) ameliorates degenerative change in a lapine surgical instability model of OA.(6) IL-1 BIOLOGY IL-1 and IL-1ra compete for binding to IL-1 receptors on the cell surface, thus providing a mechanism for titrating the cellular response.(11) The single IL-1ra gene yields four known IL-1ra transcripts derived by alternative splicing. One mRNA variant encodes a polypeptide (sIL-1ra) that is secreted by the classical pathway, while three variants predict polypeptides that lack signal peptides and thus are termed intracellular (icIL-1ra).(12-17) STUDIES OF IL-1 AND IL-1RA IN OSTEOARTHRITIC
CARTILAGE
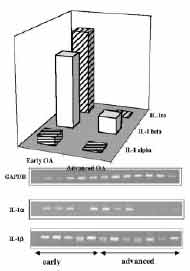
Because the IL-1 cytokines accumulate within chondrocytes in the cartilage of osteoarthritic patients early in the progression of disease, the relative expression of IL-1 and icIL- 1ra by chondrocytes may influence the localized activity of IL-1 in cartilage. In order to investigate this relationship, samples of osteoarthritic cartilage were obtained from patients undergoing knee or hip arthroplasty. Clinical data were reviewed to exclude secondary OA and inflammatory joint diseases such as rheumatoid arthritis. Cartilage was excised taking care to avoid fibrocartilage, adjacent tissue and osteophytes. Specimens of cartilage were taken from locations that macroscopically appeared to be typical in the extent of degenerative change for the preparation of histological sections. Safranin O-stained sections were examined for features such as staining intensity, cellularity, and integrity of the articular surface, and specimens were scored for OA severity using the histologicalhistochemical grading system of Mankin et al.(18) Specimens of OA cartilage were classified based on the Mankin score as either early or advanced OA. Light micrographs of safranin O-stained sections of typical early and advanced OA cartilage specimens are shown in (Figure 1).
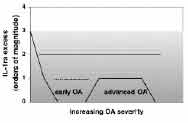
Chondrocytes were isolated from cartilage specimens for mRNA and protein analyses. IL-1α, IL-1β, and intracellular IL-1ra mRNAs were assessed by RT-PCR, and chondrocyte lysates were analyzed by ELISA for the respective proteins. IL-1α and IL-1β mRNA and protein were detected in most specimens of OA cartilage. However, in advanced OA both IL-1 agonist proteins decreased, correlating with a decrease in IL-1α and β mRNA (Figure 2). Both IL-1β and IL-1ra protein levels in chondrocyte lysates generally exceeded IL-1α levels by one order of magnitude. The exception was cases of moderate OA (grades 4-5), where antagonist levels were only slightly higher than IL-α levels (Figure 3).
Signaling through IL-1 receptors is exquisitely efficient, and binding of agonist to only a few receptors per cell is sufficient for full activation. In order to prevent IL-1 from binding to enough cell surface receptors to activate the cell, IL-1ra levels must exceed agonist levels by two to three orders of magnitude.(7) OA chondrocytes contain IL-1β, presumably in the inactive precursor form, at levels(10) times greater than those of IL-1α and cytoplasmic IL-1ra. The presence of active IL-1β converting enzyme (ICE) in human articular cartilage was recently demonstrated, with greatly increased levels in OA tissue.(19) Mature, biologically active IL-1β may be released from chondrocytes after ICE clipping. Because icIL-1ra is released together with IL-1 in response to stimulus or upon trauma or cell death, it would be better localized to antagonize IL-1 effects than the readily diffusible secreted IL-1ra. However, in osteoarthritic chondrocytes that are stimulated to release the "signal peptide-less" IL-1s, IL-1ra levels are not high enough to prevent agonist binding to cell surface receptors. Thus, IL-1 exported from OA chondrocytes may locally overwhelm inhibition by IL-1ra to promote the degenerative changes. Unconventional intracellular pathways may exist for signaling by IL-1α that is neither processed nor secreted, since pro-IL-1α is competent to bind to and activate IL-1 receptors. Signaling pathways may be activated by binding of pro-IL-1α to receptors internalized from the cell surface, or perhaps to as yet unidentified intracellular IL-1 binding proteins. Intracellular IL-1ra may also regulate intracellular signaling. IL-1ra, which is believed to be an important endogenous regulator of IL-1 signaling, was only in slight excess over IL-1α in chondrocytes from cartilage approaching the transition to advanced OA. Furthermore, antagonist levels exceede d IL-1α levels by the two orders of magnitude required to block signaling through cell surface receptors in only one case (Mankin score of 1). Particularly in these cases of moderate OA, intracellular antagonist may not be sufficiently abundant to block postulated intracellular functions of precursor IL-1α . Thus we postulate that intracellular and/or localized extracellular IL-1 signaling modulates cartilage metabolism at the critical transition from early to more advanced OA.
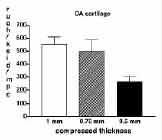
The mechanisms regulating IL-1 gene expression in OA cartilage are unknown, but IL-1 and other cytokines, various matrix degradation products such as fibronectin fragments that appear to increase catabolic activity via IL-1, and physical forces within the joint are likely culprits. In the cartilage of OA patients the large proteoglycan aggrecan, which is essential for cartilage biomechanical function, is depleted, and the additional mechanical stress under load may promote IL-1 gene expression. Recent data from this laboratory have implicated IL-1 in the anti-anabolic effects of mechanical compression in bovine articular cartilage.(20) Static mechanical compression decreases proteoglycan synthesis in human OA cartilage, as has been reported for bovine cartilage (Figure 4). Mechanical forces associated with load bearing may induce IL-1 gene expression (manuscript in preparation), trigger the release of IL-1 and icIL-1ra,(9) and perturb the cell membrane leading to clustering and internalization of IL-1 receptors.(21) It has been postulated that precursor IL-1α stored within cells is accessible to internalized IL-1 receptors and competent to activate signaling, (22) but pathways for IL-1 release and receptor internalization in "compressed" chondrocytes remain undefined. We postulate that in early OA cartilage that is weakened by the loss of aggrecan, the mechanical forces on load bearing activate IL-1 signaling in chondrocytes. SUMMARY ACKNOWLEDGEMENTS Minako Murata, MD (1,2) is a research fellow in Orthopaedic Surgery. Henry J. Mankin, MD(1) is former Chairman of Orthopaedic Surgery, Massachusetts General Hospital and Edith M. Ashley Professor of Orthopaedic Surgery, Harvard Medical School. Christine A. Towle, PhD(1,3) is an Instructor in Orthopaedic Surgery, Harvard Medical School and Assistant in Cell Biology (Orthopaedic Surgery) at MGH. (1), Orthopaedic Research Laboratories, Department of Orthopaedic Surgery, Massachusetts General Hospital and Harvard Medical School, Boston MA (2) , Current address: Department of Orthopaedic Surgery, Tokyo Women's Medical University, Tokyo, Japan (3),
Address correspondence to this author at: |
|||||||||||||||||||
|
Print Manuscript • View References • Download PDF version • Close window |